



苏钰医生的科普号
- 精选 人工耳蜗植入患者行耳聋基因诊断有什么意义?
人工耳蜗是目前解决重度感音神经性耳聋最有效的生物医学工程装置,但仍有患者耳蜗植入后效果不理想。造成不同个体其术后效果差异的影响因素除了植入年龄是主要因素外,不同基因突变导致从耳蜗到听神经范围内不同的损伤部位可能是其另外的关键因素。随着耳聋基因检测技术的不断进步,基因诊断已经成为人工耳蜗植入手术患者术前评估、术后效果预测的重要工具。以遗传性耳聋患者基因组信息为基础,对耳聋做出精准分类和诊断,可为病人设计出个体化的预防和治疗方案,实现遗传性耳聋的精准医疗。1. 不同耳聋基因突变所致内耳病变的差异与CI效果相关1.1GJB2耳聋基因突变所致耳聋GJB2基因是中国人最常见的致聋基因,常染色体隐性遗传,20%耳聋患者因此基因突变致聋,人群携带率高达11.71%(包括c.109G>A, 8.41% )。该基因编码膜缝隙连接蛋白,该通道对信号转导和物质交换起着重要作用,也是电解质、第二信使等物质在细胞间传递的重要通道,缝隙连接蛋白通道的异常可以导致耳蜗毛细胞损伤,影响听力,但由于该基因缺陷所导致病变部位仅涉及内耳毛细胞突触前结构,因而预测人工耳蜗植入效果良好,大量文献对这类人工耳蜗植入人群术后言语康复效果的研究结果很好地证明了这一推测。1.2线粒体基因突变所致的耳聋线粒体12SrRNA基因是药物性聋直接相关的责任基因,约有20%-30%的药物性聋与其相关,中国人最常见的突变位点包括A1555G、C1494T等。A1555G和C1494T的致聋机理已得到确定:A1555G或C1494T突变在12S rRNA的A区形成一个新的结合碱基对,使12SrRNA的二级结构与细菌的16SrRNA更为相似,促进了氨基糖苷类抗生素与之结合,使得带该类突变的个体在使用氨基糖苷类抗生素后会发生听力缺失。在中国各个地区统计的数据中,在非综合征性耳聋中,线粒体A1555G位点突变率为1.76%,C1494T位点突变率为0.16%。线粒体基因突变引起的药物性耳聋患者进行人工耳蜗植入的效果较理想[5, 6]。1.3内耳畸形相关耳聋内耳畸形的产生与多种因素相关。前庭水管扩大(伴或不伴IP-Ⅱ(Mondini))最为多见,约占内耳畸形患者78.2%; POU3F4基因突变所致的IP-Ⅲ内耳畸形占1.1%,其余畸形尚未明确致病基因。SLC26A4基因是常染色体隐性遗传DNFB4及Pendred综合征的责任基因,是中国第二大致聋基因,与大前庭水管综合征(Enlarged Vestibular Aqueduct Syndrome,EVAS)密切相关,23 %耳聋患者因此基因突变致病,正常人携带率为5%。大部分表现为先天性重度耳聋,一部分表现为新生儿听力筛查通过,逐渐出现程度不等迟发性或进行性耳聋;轻度的头部外伤、发热可诱发及加重耳聋。早期行基因检测发现EVAS,对一些该基因突变导致的迟发性听力损失,通过早期干预,可减缓耳聋发生。多篇文献报道SLC26A4基因突变所致大前庭水管综合征患者人工耳蜗植入,术后SIR、CAP和MAIS显著高于无突变组[3, 8]。POU3F4基因X连锁隐性遗传耳聋DFNX2的责任基因,主要临床特点为女性携带,男性发病,混合性、或进行性听力损失。CT影像表现为特征性内耳畸形:镫骨固定、内听道底异常扩大,蜗轴缺如、耳蜗底转骨质缺如,耳蜗与内听道相通。对于重度极重度感音神经性耳聋的DFNX2患者,人工耳蜗植入是唯一选择。但由于耳蜗结构异常,可能导致术中电极误入内听道、术中井喷和术后脑脊液漏等风险,因而手术具有一定难度[9]。对POU3F4基因所致的DFNX2耳聋患者进行人工耳蜗植入,多数术后听力康复效果显示患者听力恢复情况良好,但整体言语识别率、听力和言语能力低于耳蜗结构正常儿童人工耳蜗植入的平均水平[11],也有文章报道康复效果可能与基因突变的类型相关。1.4听神经病所致耳聋听神经病谱系障碍(Auditory neuropathy spectrum disorder,ANSD )又称听神经病 (Auditory neuropathy,AN )。听神经病的诊断标准为毛细胞的功能正常——耳声发射(OAE)和/或耳蜗微音器电位(CM)可引出,而听神经功能异常——听性脑干反应(ABR)异常或全部消失,同时多可伴有中枢或周围神经病变。40%的听神经病与遗传因素相关。目前已发现20个与听神经病发病相关的基因,包括与非综合征型听神经病相关的OTOF、PJVK、DIAPH3、SLC19A2、SLC17A8、12SrRNA,与综合征型听神经病相关的PMP22、NF-L、MPZ、NDRG1、GJB1、GJB3、OPA1、TMEM126A、FXN、TIMM8A、WFS1、AIFM1[13]基因。有报道线粒体突变12rRNA(T1095C)[14]和MTND4 (11778mtDNA)[15]与听神经病相关,但仍需更多的病例数及功能学验证来证实。不同的基因突变导致的听损部位不同,根据听损部位可分为听神经病变型(突触后型)(PJVK、MPZ),听突触病变型(突触及突触前型)(OTOF、DIAPH3、SLC19A2、SLC17A8)和非特异性(线粒体相关)三类。基于不同基因突变,引发不同部位听损病理改变及人工耳蜗作用原理,不同在人工耳蜗植入术后预后不同。OTOF基因,又称DFNB9。根据已有数据听神经病谱障碍中的OTOF基因突变患者,无一例外地从人工耳蜗植入中获益,这是因为Otoferlin 蛋白表达异常的患者病变处于内毛细胞或内毛细胞与听神经形成的传入突触,而听神经纤维正常,因此这类患者进行人工耳蜗植入后效果比病变位于听神经Ⅰ型听神经病患者的效果更好。有报道听神经病谱障碍基因中的4个基因OPA1、DIAPH3、AIFM1、DFNB59突变,由于其直接对螺旋神经节细胞造成的损害,接受人工耳蜗植入的患者术后言语感知能力明显比其他患者要差。同时也有报道携带OPA1基因患者术后言语识别和ABR均有提高,人工耳蜗植入效果显著。Wu CC等人对12个人工耳蜗植入效果差的患者进行了高通量测序发现有4例为PCDH15基因致病突变, 3 例患者是(PJVK)DFNB59基因突变致病。目前研究认为听突触病变型(突触OTOF及突触前型DIAPH3)及线粒体相关病变(OPA1)患者人工耳蜗预期植入效果较好。明确AN患者的遗传学病因结合听觉电生理检查,对于行人工耳蜗植入术前评估术后康复具有积极的指导意义。1.5其他耳聋基因突变所致耳聋人工耳蜗植入也被认为能为DFNA9 (COCH基因) 、MYO15A、MYO6、MYO7A、TECTA、TMPRSS3、TMC1、和ACTG1等基因突变患者提供比较好的疗效, 有报道发现TIMM8A基因植入效果差。TMPRSS3 基因编码II型跨膜丝氨酸蛋白酶,该基因突变可引起:语前聋ARNSHL(DFNB10):先天性重度-极重度感音神经性耳聋;语后聋ARNSHL (DFNB8):迟发性,中高频听力下降为主,进行性。TMPRSS3表达在内耳毛细胞、血管纹及螺旋神经节细胞,由于其在螺旋神经节细胞中明显表达,在最初鉴定该基因时不建议患者行人工耳蜗植入术,我们和国外的数个报道均显示TMPRSS3耳聋患者行人工耳蜗植入效果良好。另外,还有一些未知基因尚未发现,有待进一步探索。2. 基因检测有助综合征性耳聋的精准诊断、预测人工耳蜗植入效果目前已报道的综合征型耳聋(Syndromic hearing loss, SHL)有400 多种,大多数发病率低,有些仅见个别报道,临床上较常见的有Waardenburg 综合征(MITF、PAX3、SOX10、SNAI2、ENDRB 、EDN3)、Van der Hoeve综合征(COL1A1、COL1A2)、Usher综合征(MYO7A、 USH1C、CDH23、 PCDH15、SANS、 CIB2、 USH2A、VLGR1、WHRN、 CLRN1、PDZD7)、CHARGE综合征(SEMA3E、CHD7)、Alport综合征(COL4A3、COL4A4、COL4A5、COL4A6)等等。综合征型耳聋基因涉及多个系统,涉及的致病基因繁多,却可以导致很多共同的临床表型,它们之间可能存在着潜在的共同通路和相互作用网络。对SHL遗传基础及发病机制的系统研究变得十分困难。但随着医学科技进步和社会经济发展,一些严重的综合征患者的生存率越来越高,越来越多的SHL患者及家庭要求通过人工耳蜗植入的方式提高听力,但患者是否适合手术、能否耐受手术,并最终从CI植入获益,给耳外科医生提出了更高的要求。Waardenburg 综合征(WS)又称听力色素综合征,以感音神经性耳聋、皮肤低色素(白化病)、白额发或早白发、虹膜异色为主要临床症状。WS 是最常见的综合征型耳聋之一,人群发病率为1/42000,聋哑人群中发病率为0.9%-2.8%,占先天性耳聋的2%-5%。依据不同的伴随表型,可将WS 分为4 种亚型(WS1-4),最常见的为WS1 和WS2。不同的致病基因与不同的WS 亚型相关联,PAX3 基因突变可导致WS1 和WS3,MITF 基因突变导致WS2A,SNAI2 基因突变导致WS2D,SOX10 突变导致WS2E。SOX10、EDN3 和EDNRB 基因突变可造成WS4。国内相关研究表明中国人群WS 主要致病基因为PAX3、MITF 和SOX10。WS 相关基因的突变会造成NC 发育异常,导致黑素细胞发育不良,引起黑色素合成减少,表现出皮肤、毛发、虹膜低色素等临床症状。血管纹是内耳耳蜗结构之一,在内淋巴生成过程中起重要作用。研究显示,黑素细胞发育不良会引起血管纹中黑素细胞源性的中间细胞缺乏进而造成柯蒂氏器(Corti)退化变性,最终导致感音神经性耳聋。根据目前报道最多的是Waardenburg综合征1型和2型合并极重度感音神经性聋患者人工耳蜗植入,术后听觉效果与一般的耳蜗植入患者无显著差异。CHARGE综合征患者中80-100%病例伴有耳聋,其中50%为重度-极重度耳聋,常伴有内耳畸形、内听道狭窄、听神经缺如,亦可伴有中耳畸形、面神经畸形等。CHARGE 综合征患者行人工耳蜗植入术前需MRI评估听神经是否发育不良或缺如;术中需注意面神经走行异常,遮挡圆窗龛等畸形;人工耳蜗植入术后听力康复和言语发育的疗效存在较大个体差异。Annemarie等人报道了10例确诊为CHD7导致的CHARGE综合征患者人工耳蜗植入术后随访5年,其中2例获得满意言语康复效果,2例需配合手语进行一定程度的言语交流,余6例不能进行言语交流[28]。CHARGE综合征患儿的生长发育迟缓、脑白质发育不良、不同程度的自闭症等是影响术后言语康复效果的重要因素。目前已知的CHARGE综合征致病基因为CHD7和SEMA3E,尚无文献报道基因突变的类型、位置、大小与人工耳蜗术后康复效果相关。还有一些更为少见的SHL,如由我国袁永一教授首先发现并鉴定ATP6V1B2 基因为其发病的责任基因的耳聋甲发育不全综合征(DDOD),其主要临床表现为先天性耳聋、指/趾甲发育不全、指骨发育不良、锥形牙齿等。目前国际上有四例DDOD患者接受了人工耳蜗植入,其言语康复效果差于同龄植入的SNHL患者,可能与DDOD综合征患者存在学习记忆障碍相关。事实上,SHL极为复杂,有一些少见但全身情况更为复杂的SHL。如伴有心脏缺陷的SHL,包括与KCNQ1基因及KCNE1基因相关的聋心综合征(Jervell&Lange Nielsen 综合征)(心源性猝死)、与PTPN11基因突变相关的Noonan综合征(凝血功能异常);与神经及神经肌肉系统关系密切的SHL,如与TWINKLE、CLPP、LARS2、C10ORF2、HSB17B4基因相关的Perrault综合征、与MERLIN基因相关的Ⅱ型神经纤维瘤病;还有大量伴有代谢性疾病、内分泌疾病、肾病、眼、口、皮肤的SHL等等,需在术前充分准备,并对预后有所预期。基因诊断可能实现综合征性耳聋的精准诊断,提示患者疾病可能包含的多器官异常,为CI候选者的全身状况和耐受能力的评估提供客观数据,实践多学科协作、确保围手术期安全,同时预测人工耳蜗的效果,决策人工耳蜗适应症范围。3. 耳聋基因诊断对CI手术时机、侧别选择及电极选择具有指导意义GJB2基因 和SLC26A4基因是中国人最为常见的耳聋基因,研究也较为透彻。这两种基因突变所致的耳聋,在人工耳蜗植入的侧别选择方面做听力差的一侧即可获得确定的疗效,而对一些原因不明的极重度感音神经性耳聋患者,做电生理能够引出反射的一侧,似乎更加保险。还有一些渐进性、迟发型听力损失的患者,如SLC26A4、DFNA5、TECTA等基因突变患者可根据听力损失进展情况,个体化选择听力干预手段及人工耳蜗植入时间。电极选择方面,对于有低频残余听力患者,理论上可以选择声电联合刺激模式,但是对于基因检测显示耳蜗及螺旋神经节上均有表达的基因,如:TMPRSS3 基因、 PCDH15基因突变,即使术后表现良好也可能会出现进行性听力减退,人工耳蜗植入后需根据残余听力进展情况对耳蜗刺激模式进行调整。对于内耳畸形患者,需根据内耳发育情况结合基因检测选择电极。如POU3F4、COL4A6基因突变所致的不全分隔 Ⅲ型内耳畸形,需选择短电极直电极,使电极贴耳蜗外侧壁。而耳蜗形态正常患者,除直电极外还可选择抱轴弯电极或中位电极。2002年起,解放军总医院聋病分子诊断中心采用全频谱覆盖耳聋基因筛查的策略开展了全国范围内的耳聋流行病学研究,目前已收集耳聋病例21000余例,进行了系统性耳聋基因致病性及流行病学分析,绘制了中国人耳聋基因突变的基因谱和突变谱。常规开展耳聋基因诊断及产前诊断,在耳聋的三级预防工作中发挥了重要作用,具有重要意义。目前解放军总医院耳蜗中心的人工耳蜗植入的例次已超过4300例。综上,耳聋基因不同类型突变导致了内耳病变的差异,同一基因不同突变模式,同样会影响人工耳蜗植入后的康复效果,耳聋基因诊断对于人工耳蜗植入效果的预测及评估的重要性不言而喻,它将是实现遗传性耳聋精准治疗的重要工具。(苏钰 戴朴 解放军总医院)
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科4525人已读 - 精选 好消息,先天性结构畸形有免费救助啦!
先天性结构畸形救助项目方案为减少先天性结构畸形所致残疾,推进健康扶贫工程,国家卫生计生委妇幼司、中国出生缺陷干预救助基金会(以下简称基金会)于2017年启动实施先天性结构畸形救助项目。项目主要针对发病率相对较高、有成熟干预技术、治疗效果好的先天性结构畸形,为患儿提供医疗费用补助,减轻患儿家庭医疗负担。为确保项目顺利实施,特制订本方案。一、项目目标(一)普及先天性结构畸形防治知识,提高公众优生意识。(二)减轻贫困患儿家庭医疗负担,促进先天性结构畸形早诊断、早治疗,减少先天性结构畸形所致残疾。二、项目内容(一)以先天性结构畸形为重点,开展出生缺陷防治社会宣传和健康教育。(二)开展人员培训和业务指导。(三)为符合救助条件的患儿提供医疗费用补助。三、救助对象及补助标准(一)救助对象。申请救助的患儿需同时满足下列条件:1.临床诊断患有下列6类先天性结构畸形疾病:①神经系统先天性畸形;②消化系统先天性畸形;③泌尿系统及生殖器官先天性畸形;④肌肉骨骼系统先天性畸形;⑤呼吸系统先天性畸形;⑥五官严重先天性结构畸形。2.年龄18周岁以下(含)。3.家庭经济困难,能够提供低保证、低收入证、建档立卡贫困户证明或村(居)委会等开具的贫困证明(附2)。4.在项目定点医疗机构接受诊断、手术、治疗和康复。5.医疗费用自付部分超过3000元(含)。(二)医疗费用补助范围。药费、床位费、诊查费、检查费、放射费、检验费、治疗费、手术费、输血费、护理费、材料费、输氧费等。(三)补助标准。对患儿申请救助日期的上一年度1月1日(含)之后,在定点医疗机构的诊断、手术、治疗和康复医疗费用给予补助。项目根据患儿医疗费用报销之后的自付部分,一次性给予3000元-30000元补助。每位患儿具体补助标准如下:1.自付部分超过3000元(含)、小于4000元的,补助额度为3000元。2.自付部分超过4000元(含)的,按自付费用的75%予以补助,最高补助额度为30000元(含)。对同一患儿同一疾病分次申请救助的,或同一患儿不同疾病分别申请救助的,项目只补助一 (更详细的内容,可搜索中国出生缺陷干预救助基金会官网自行学习)先天性结构畸形申请表和贫困证明可在中国出生缺陷干预救助基金会官网下载专区下载。需要救助的耳鼻咽喉患者可于解放军总医院耳鼻咽喉头颈外科 苏钰副主任医生的门诊就诊咨询手术及费用减免事宜 (门诊时间每周三下午,301医院门诊大楼10层B区3诊室)。
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科4601人已读 - 精选 新生儿八种耳廓畸形耽误不得
新生儿耳廓畸形发病率很高。小耳畸形患者需要7岁以后手术进行耳再造,也有一些轻度的耳廓变形可以自愈,下面图示的这八种耳廓形态异常则需要佩戴耳矫形器,进行早期的非手术矫治,越早矫治效果越好。新生儿耳畸形无创、非手术最佳矫治时间为出生7至30天,发现后及时就诊尽早安装矫治用耳模型。如果是 30天内的婴儿,治疗2周就可以到达95%的治愈率。大于30天的婴儿,治疗疗程大约在2-6周。在治疗周期内请按照医生要求,定期回访。错过30天的治疗窗时间,治愈率会随之下降;大于6个月的婴儿基本就不能进行无创矫治了。目前应用的Earwell爱韦尔外耳廓矫正器(原装进口)是美国、加拿大、欧洲、新加坡等发达国家临床使用最广泛的治疗性外耳廓畸形矫正系统,矫正率达95%,大多数婴儿无需再进行手术矫正。矫正器的材质为全球顶级医用聚氨酯类热塑性弹性体材料(TPE),供应商为3M和Henkel公司,并通过多项生物相容、粘膜刺激、致敏、毒理等生物学评价检测合格,材料内不含乳胶、增塑剂(DEHP) 、苯二甲酸酯,双酚And 8(BPA)等致敏或致癌成份,对婴儿皮肤无刺激、无致敏、无毒性。获美国FDA、CE认证,国际发明专利—200880108740X。 如有疑问请留言咨询
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科2.1万人已读 - 精选 并发于希林氏病的内淋巴囊肿瘤的诊断与治疗
内淋巴囊肿瘤(Endolymphatic sactumor,ELST) 是起源于颞骨的肿瘤,根据其组织来源于又称内淋巴囊的低度恶性腺癌(Low gradeadenocarcinoma of probably endolymphatic sac)或 Hefner肿瘤。ELST极少见,可单独发生,多伴发于Von-Hippel-Lindau综合征(Von-Hippel-Lindau syndrome ,VHL病,又称希林病)。ELST患者病程较长,因肿瘤侵袭颞骨岩部,首发症状主要有听力下降、耳鸣、眩晕等,随着肿瘤体积的增大,可伸入脑桥小脑角或相邻结构,引起相应占位效应,常见受累的脑神经为第8颅神经(面神经),其次为第9 、10 、12、5对脑神经, 部分患者可见肿瘤侵犯鼓膜或进入外耳道。尽管其恶性程度较低,但其位于侧颅底,病变部位深在,紧邻重要神经血管及后颅窝,术前难以确诊,手术难度较大,术后并发症较多、较重,因此有必要了解其临床和影像学特点以及分子病因以便进行正确的诊治。VHL最初在1895年由德国眼科医生vonHippel 首先发现了视网膜血管母细胞瘤患者具有家族特性。1926年瑞典眼科医生ArvidLindau发现视网膜血管母细胞瘤并发小脑血管瘤及腹腔脏器病变的病例,因而将脑-眼-内脏血管瘤综合症称为VHL病。1964年,Melon和Rosen总结了多篇临床报告,将中枢神经系统(CNS)血管母细胞瘤合并肾脏或胰腺囊肿、嗜铬细胞瘤、肾癌以及外皮囊腺瘤等疾病正式命名为“von Hippel Lindau综合征”,简称VHL综合征。VonHippel-Lindau(VHL)综合征是一种常染色体显性遗传病。发病率约1/36,000 -1/53,000,发病年龄为18-30岁,60岁有95%的外显率。10%的病例为儿童期发病,主要特征性病变为视网膜和中枢神经系统血管母细胞瘤和嗜铬细胞瘤。临床诊断标准:对于家族中有视网膜血管瘤或中枢神经系统成血管瘤遗传病史的患者只要存在视网膜血管瘤、中枢神经系统成血管瘤或实质性脏器损害(肾癌、胰腺多发性囊肿或肿瘤、嗜铬细胞瘤、附睾乳头状囊腺瘤)中的任何一个,即可诊断。对于无家族史的散发性患者具备两个以上 (含两个)血管瘤或一个血管瘤和一个实质性脏器肿瘤亦可诊断。 不同家族及同一家族不同患病个体可有不同的临床表现,最初症状往往不只出现在一个器官,不发生在固定的年龄段,而且疾病的严重程度差异很大,很多家族成员只有相对无害的表现,而另一些成员则病情很重。另外,各种肿瘤在不同家族、不同人种中的发生率差异很大:法国人易患神经系统肿瘤,德国人易患嗜铬细胞瘤,日本人更易患肾肿瘤。患病率和致死率最高的伴发症状为肾囊肿和肾癌,中枢神经系统血管瘤次之。 在VHL认识的进程中,内脏表现重要性的发现,新的特异性中枢神经系统表现(内淋巴囊肿瘤)及附件肿瘤的认识,尤其是近年来,VHL基因特征、功能、突变检测方面的进步,为患者的临床诊治带来巨大变化。内淋巴囊肿瘤(ELST) ELST是一种罕见的神经外胚层颞骨肿瘤。自从1898年Treiter第一次报道了颞骨乳头状腺瘤后,一个世纪以来,对于ELST的分类就一直存在争议。关于肿瘤的起源有各种假说:外耳道耵聍腺、异位的小涎腺、中耳腺体、中耳粘膜上皮、脉络丛乳头状瘤等等。命名方面也五花八门:包括桥小脑角耵聍腺瘤、腺样囊性腺瘤、多形性腺瘤、颞骨腺癌、侵袭性乳头状中耳颞骨肿瘤等等。直到1984年,Hassard和MacDougall相继报道了内淋巴囊起源的颞骨肿瘤的病例,1989年Heffner证实了肿瘤内淋巴囊起源这一假说,内淋巴囊肿瘤这一命名才得到广泛的承认。 尽管1904年起就有VHL并发ELST报道,且有2-11%VHL病患者并发ELST。但是近十年左右,ELST才作为VHL的临床表现之一被认识。事实上,ELST罕见出现在非VHL病的患者中, VHL合并ELST的患者约有30%为双侧发病,95%伴发听力减退,92%伴发耳鸣,62%伴发眩晕及平衡失调,29%有外耳道胀满感,8%伴发面神经受损症状。听力减退通常会在早期出现(平均年龄22岁),约43%为急性发病。临床症状的轻重与肿瘤的大小并不成正比。Lonser通过对VHL合并微小ELST的患者颞骨组织学研究后认为,急性的内淋巴出血及其造成的炎症可能为急性听力减退的主要原因,而水肿则可以解释该病梅尼埃样症状(耳聋、耳鸣及眩晕)的出现。影像学检查 影像学检查对ELST的发现至关重要。约11%-16%的患者是通过MRI或CT检查发现的。Mukherji等最早对ELST的影像学特点进行了研究,认为ELST的CT表现具有特征性,肿瘤中心位于内听道和乙状窦之间岩骨后缘的前庭导水管区域, 直径<2cm的肿瘤表现为岩骨迷路后前庭导水管外孔区软组织肿块和前庭导水管骨质破坏, 随肿瘤增大, 可累及范围包括鼓室、迷路骨质等内耳周围结构及颈静脉孔区, 骨质破坏多为蜂窝状、溶骨样。肿瘤内有点片状及针状高密度骨质, 代表活动性骨破坏结果,反映肿瘤生长缓慢,肿瘤后缘见薄层钙化缘是重要的诊断依据。在组织学上肿瘤的中心包含骨针和蜂窝网状骨性结构,而EI ST本身不产生骨样组织。因此,HRCT上所显示的高密度骨影可能是肿瘤浸润包埋造成的残留骨形成包绕的钙化边。 MRI上信号混杂,较特异性的表现有,不增强的T1WI有增加的信号密度,各种肿瘤内病灶显示为斑点样;T2WI 有散在信号加强区和流空现象,强化后出现标志性的高信号。治疗 ELST以手术彻底切除为主要治疗手段。凡MRI发现肿瘤或出血但仍有听力的患者,均需要手术治疗以防病情恶化。如果听力丧失同时伴有其他神经系统症状,也应手术治疗。但如果患者有ELST的相关症状,而影像学检查未见异常,是否应行手术以防止听力丧失及其他症状,尚需更多的临床研究。手术入路的选择方面,因原发病灶位于迷路后故一般采用耳后进路,病变范围广泛者可行耳颈联合进路。手术先行乳突切除并切除乳突后方骨壁,显露病变及乙状窦和后颅窝硬脑膜,外耳道未受累者可保留骨性外耳道后壁,病变侵犯外耳道时则应予切除并封闭外耳道,必要时可行颞骨次全切除;面神经未受累时应保留其骨管或神经,如面神经受累则应予切除,并取耳大神经进行神经移植或行面神经-舌下神经吻合;病变侵犯硬脑膜不易分离时应切除受累硬脑膜,用颞肌筋膜修补,术腔可用颞肌胸锁乳突肌或脂肪充填。肿瘤多为颈外动脉及小脑前下动脉供血,术前应行血管栓塞,以减少术中出血,缩短手术时间和减少并发症。手术切除不彻底时可复发, 但未见肿瘤转移的报道。常规放射治疗和化学治疗对于术后残留肿瘤无根除作用,但γ刀治疗对本病有效。VHL基因的研究进展VHL基因是一个典型的抑癌基因。1993年,Latif等发现了该基因,将其定位于3p 25-26,并通过位置克隆的方法分离成功。VHL基因全长4.5kb,分布在约占20 kb的DNA空间。该基因含3个外显子,VHL基因编码含有213个氨基酸、分子量为30000的蛋白质,又称VHL 蛋白。该蛋白负性调控血管内皮生长因子(VEGF)mRNA的表达。VHL基因突变可造成该蛋白功能丧失,VEGF表达升高而发生富含血管的血管母细胞瘤。 最初VHL家系基因突变的检测率约为39-75%,通过实验方法的改进,Southernblot 、荧光原位杂交(FISH)及直接测序的使用,目前VHL家系基因突变的检测率几近100%。 迄今为止,没有有效的药物来预防或治疗有临床表现VHL患者。因为大量的肿瘤不能手术切除,而且复发率高。近期pVHL的功能研究证实pVHL在调节VEGP和血管发生中的重要作用,这可能将对VHL及相关疾病的治疗提供新的策略。在体外,VEGF对新生的肿瘤血管来讲是重要的生存因子,撤退VEGF能够导致出血、坏死及血管退化。最近的研究,当使用血管生成抑制药物后,大鼠VHL肿瘤的生长率明显抑制,血管生成率和体积均明显下降。因而,目前大量的实验集中在使用特异药物阻断VEGF,以期控制或抑制CNS和视网膜血管母细胞瘤的生长。VHL基因突变的人群携带率估计为3/10万左右,外显率接近100%。其遗传特征为常染色体显性方式,子女有50%机率发病,故对其子女也应严密随访。结论 总之,ELST是一种极为少见的颞骨岩部或可扩展至桥小脑角及周边结构的肿瘤,极易伴发于VHL病患者,分子诊断对可疑患者、高危患者的症状前诊断非常重要,对检测出的携带者进行密切随访,可以早期发现肿瘤、早期治疗。
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科4117人已读 - 医学科普 耳廓畸形能治愈么?如何治疗?
耳廓畸形可以治愈。但治疗方法与耳畸形的严重程度直接相关。 耳廓形态畸形(耳廓的结构都在,只是形态异常,俗称的招风耳、杯状耳、猿耳、垂耳等都属于这一范畴),可以在新生儿出生后最佳时间0-21天通过使用耳廓无创矫正器来矫正,我们的经验是出生后1个月以后才开始矫正的孩子,也会有效,相较早期来看,时间长,效果也大打折扣。如果错过这个时间则需要等到孩子五六岁时再做手术来完成矫形,实际上有一些类型的形态畸形,手术设计非常困难,手术也很难达到很理想的效果,当然还存在全身麻醉、感染等手术风险。 对于有组织缺损较多的小耳畸形,一般要等到7岁左右,行耳再造手术恢复外形,同时在出生后要尽早联系耳畸形专业耳鼻喉科大夫评估听力和耳道情况。
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科1872人已读 - 医学科普 新生儿耳廓畸形诊断、最佳治疗时机及预后
解放军总医院耳鼻喉科 苏钰 新生儿耳廓畸形发病率很高。伴有组织缺损的小耳畸形患者需要7岁以后手术进行耳再造。一些轻度的耳廓变形可以自愈,还有一些耳廓形态异常则需要在新生儿期佩戴耳矫形器,进行早期的非手术矫治,越早矫治效果越好。由于一些习俗和家长认识不足,导致很多新生儿都错失了宝贵的黄金治疗期,即使后期矫正效果也不理想。耳廓畸形不仅仅会影响美观,而且随着宝宝的成长会产生心理影响,从而表现出自卑、焦虑、社交困难及消极情绪。 1. 那么什么样的耳畸形需要接受矫形治疗呢? 答:无皮肤及软骨缺损的耳廓变形,如垂耳,杯形耳,招风耳,Stahl’s耳,环缩耳, conchal crus耳,隐耳等 。其中很多耳部畸形是混合存在的(含2种或以上的畸形)。 2.新生儿耳矫形的原理是什么呢? 答:新生儿出生时体内血液中含有一定浓度的母体雌激素,新生儿的耳软骨中玻尿酸的浓度水平也较平常高,此时宝宝耳朵非常柔软可塑性好,正是佩戴耳矫形器进行无创,无痛非手术矫正形态耳畸形的黄金期。新生儿体内的母体雌激素在出生后72小时达到峰值,6周后降至正常水平。 3.新生儿耳廓畸形矫形最佳矫治时机是什么时候呢? 答:早期干预,越早矫正效果越好,佩戴耳矫形器的时间也相对短,越晚效果效果越差。新生儿耳廓畸形的最佳矫治时间为出生后5天至1个月以内。出生超过90天,除隐耳外大多数形态耳畸形基本错过了矫正的最佳时期。 4.耳矫形器是什么样子的呢? 答:我们目前使用的是Earwell爱韦尔外耳廓矫正器(美国原装进口)。材质为全球顶级医用聚氨酯类热塑性弹性体材料(TPE),供应商为3M和Henkel公司,对婴儿皮肤无刺激、无致敏、无毒性。获美国FDA、CE认证,国际发明专利—200880108740X。 5.耳矫形器治疗效果怎么样呢? 答:对于符合条件的新生儿,出生 30天内,治疗2周就可以到达95%的治愈率。大于30天的新生儿,治疗疗程大约在2-6周。(附部分治疗前后图例) 6.耳矫形器治疗会有并发症吗? 答:绝大部分的新生儿都能顺利的完成矫治,少数治疗过程中可能会出现轻微的局部压伤,一般清洁处理后3-4天就可以自愈,不影响矫治也不会留疤。 7.出现这种情况到哪里进行矫治呢? 答:解放军总医院耳鼻喉科是国内最早开展此项治疗的单位之一。苏钰副主任医师每周三全天门诊(门诊楼10层B区3诊室),或在苏钰好大夫网站提前预约咨询。
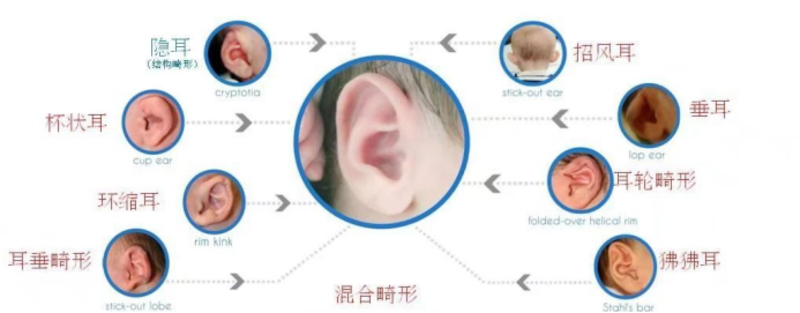 苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科9566人已读
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科9566人已读 - 医学科普 新生儿耳廓畸形的早期矫正
我科室通过近两年的临床观察,新生儿早期佩戴Earwell耳矫形器,矫正形态耳畸形取得了良好的效果(附矫正前后对照图片)。 新生儿出生时体内血液中含有一定浓度的母体雌激素,新生儿的耳软骨中玻尿酸的浓度水平也较平常高,此时宝宝耳朵非常柔软可塑性好,正是佩戴耳矫形器进行无创,无痛非手术矫正形态耳畸形的黄金期。新生儿体内的母体雌激素在出生后72小时达到峰值,6周后降至正常水平。出生超过90天,除隐耳外大多数形态耳畸形基本错过了矫正的最佳时期。 所以早期干预越早矫正效果越好,佩戴耳矫形器的时间也相对短,越晚效果效果越差,据美国临床验证表明出生2周的新生儿通过佩戴耳矫形器矫正形态耳畸形成功率可达95%。因而新生儿耳廓畸形的最佳矫治时间为出生后5天至1个月以内。 那么什么样的耳畸形需要矫正呢?是否可以通过等待自愈或者家人帮忙抻拉就能恢复正常呢?由于一些习俗和家长认识不足,导致大多数新生儿都错失了宝贵的黄金治疗期,即使后期矫正效果也不理想。爱美之心人皆有之,耳廓畸形不仅仅会影响美观,而且随着宝宝的成长会产生心理影响,有证据表明有90.3%的患儿不愿谈及畸形耳。患有面部畸形的儿童,常常遭到玩伴的嘲笑、戏弄,从而表现出自卑、焦虑、社交困难及消极情绪。 据统计,新生儿发生形态耳畸形的概率中国局部地区发生率为50%,日本新生儿统计发生率是55,2%,美国新生儿统计发生率20-30%,由此可见东方人的发病率高于西方人。其中仅有30%的轻度畸形耳可以自行改善,剩余70%会维持原样或更加严重,严重耳畸形的患儿不得不在4-6岁时接受外科手术矫正。事实上,外科手术也常常不能达到理想效果。 新生儿形态耳畸形需要在宝宝出生后即可检查,鉴别,发现耳朵形态异常应及时就诊,在黄金期内矫正取得满意的矫正效果。避免错失矫正良机,不给宝宝的成长留遗憾。请家长及时带患儿就诊治疗。 主诊专家:苏钰 副主任医师 出诊地址:301医院 中国人民解放军总医院门诊10层B区 北京市海淀区复兴路28号 出诊时间:周三全天 矫正时间:周二下午
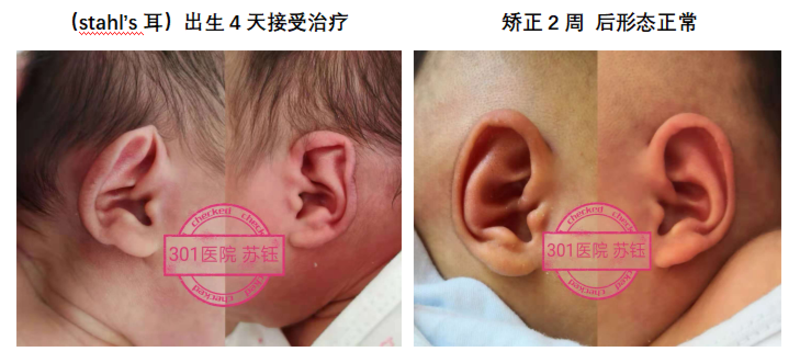 苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科2677人已读
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科2677人已读 - 案例 耳部整形
治疗前据统计,新生儿发生形态耳畸形的概率中国局部地区发生率为50%,日本新生儿统计发生率是55,2%,美国新生儿统计发生率20-30%,由此可见东方人的发病率高于西方人。其中仅有30%的轻度畸形耳可以自行改善,剩余70%会维持原样或更加严重,严重耳畸形的患儿不得不在4-6岁时接受外科手术矫正。事实上,外科手术也常常不能达到理想效果。所以发现耳朵形态异常应及时就诊,在黄金期内(出生后5天至1个月以内)矫正,效果会比较理想。这个宝宝是严重垂耳+隐耳,在出生29天后开始接受矫正,这是矫正5周效果,基本完全恢复正常。治疗后治疗后35天据统计,新生儿发生形态耳畸形的概率中国局部地区发生率为50%,日本新生儿统计发生率是55,2%,美国新生儿统计发生率20-30%,由此可见东方人的发病率高于西方人。其中仅有30%的轻度畸形耳可以自行改善,剩余70%会维持原样或更加严重,严重耳畸形的患儿不得不在4-6岁时接受外科手术矫正。事实上,外科手术也常常不能达到理想效果。所以发现耳朵形态异常应及时就诊,在黄金期内(出生后5天至1个月以内)矫正,效果会比较理想。这个宝宝是严重垂耳+隐耳,在出生29天后开始接受矫正,这是矫正5周效果,基本完全恢复正常。
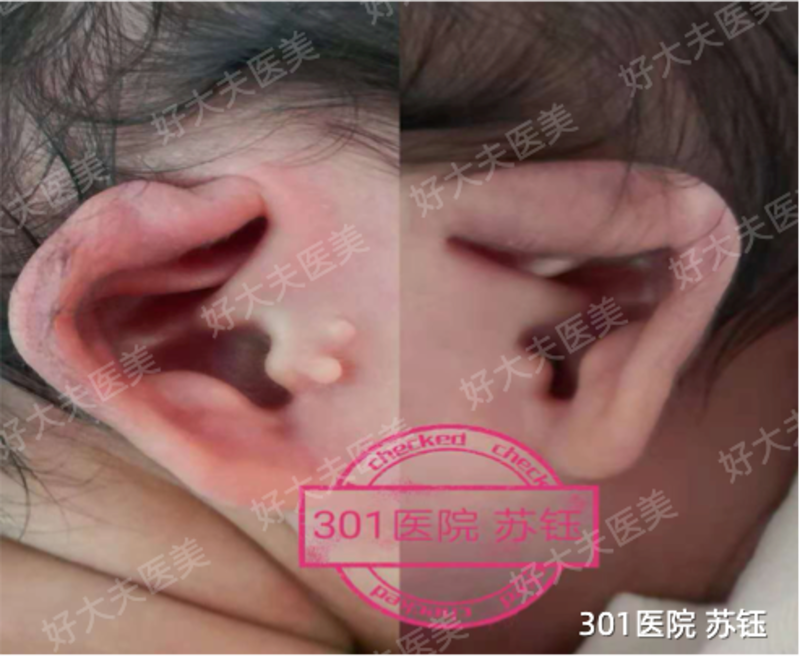 苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科164人已读
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科164人已读 - 案例 耳部整形
治疗前新生儿形态耳畸形需要在宝宝出生后即可检查,鉴别,发现耳朵形态异常应及时就诊,在黄金期内矫正取得满意的矫正效果。避免错失矫正良机,不给宝宝的成长留遗憾。请家长及时带患儿就诊治疗。案例中这个宝宝是杯状耳,出生4天开始矫正,这是佩戴1周的效果,基本上已经完全复原。治疗后治疗后7天新生儿形态耳畸形需要在宝宝出生后即可检查,鉴别,发现耳朵形态异常应及时就诊,在黄金期内矫正取得满意的矫正效果。避免错失矫正良机,不给宝宝的成长留遗憾。请家长及时带患儿就诊治疗。案例中这个宝宝是杯状耳,出生4天开始矫正,这是佩戴1周的效果,基本上已经完全复原。
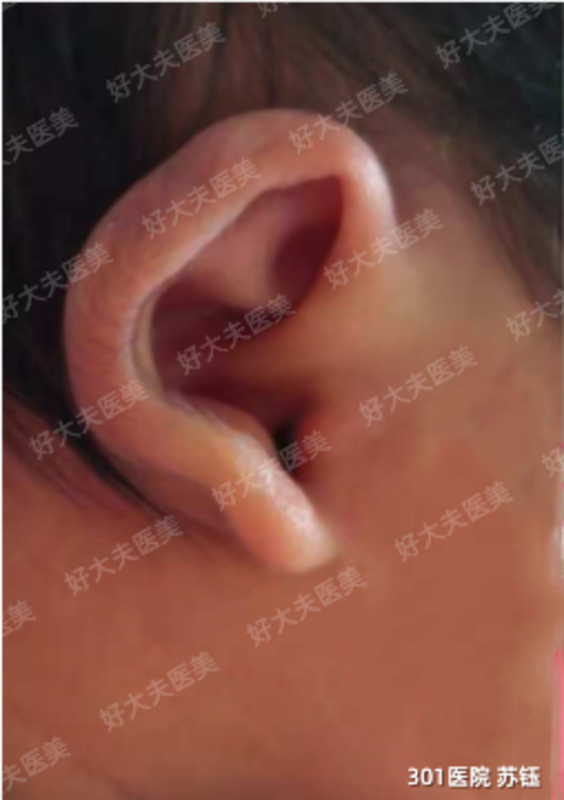 苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科53人已读
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科53人已读 - 案例 耳部整形
治疗前新生儿出生后72小时到6周期间,因为血液中含有一定浓度的母体雌激素,耳软骨中玻尿酸的浓度水平也高,因此宝宝耳朵非常柔软,可塑性好,正是佩戴耳矫形器进行无创,无痛非手术矫正形态耳畸形的黄金期。早期佩戴Earwell耳矫形器,矫正形态耳畸形可以取得良好的效果。这个宝宝是耳轮畸形,出生7天开始接受矫正。这是矫正2周后的效果,耳轮已基本恢复正常。治疗后治疗后14天新生儿出生后72小时到6周期间,因为血液中含有一定浓度的母体雌激素,耳软骨中玻尿酸的浓度水平也高,因此宝宝耳朵非常柔软,可塑性好,正是佩戴耳矫形器进行无创,无痛非手术矫正形态耳畸形的黄金期。早期佩戴Earwell耳矫形器,矫正形态耳畸形可以取得良好的效果。这个宝宝是耳轮畸形,出生7天开始接受矫正。这是矫正2周后的效果,耳轮已基本恢复正常。
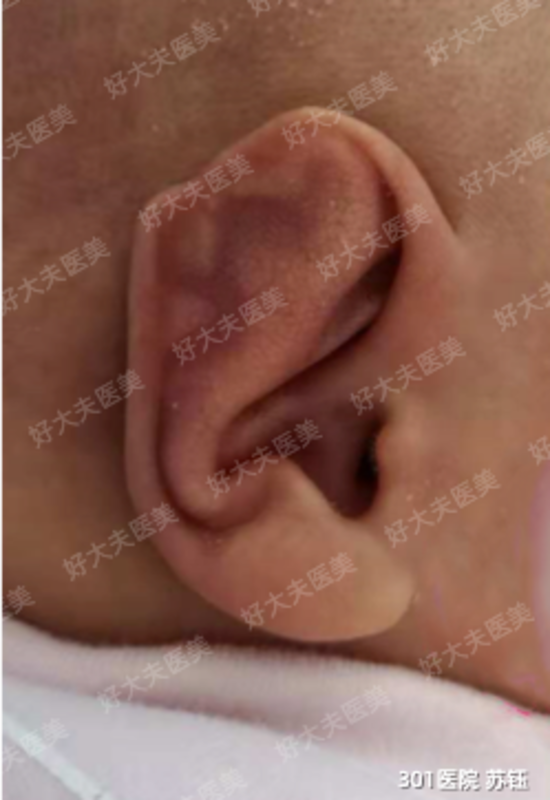 苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科57人已读
苏钰 副主任医师 中国人民解放军总医院第一医学中心 耳鼻咽喉头颈外科57人已读

苏钰副主任医师
中国人民解放军总医院第一医学中心耳鼻咽喉头颈外科
